
Rob Jensen, Ph.D.
Department of Cell Biology
Johns Hopkins University School of Medicine
725 N. Wolfe Street, 114 WBSB
Baltimore, MD 21205
Academic Titles
Professor EmeritusResearch Topic
The dynamics of mitochondria and their DNA in yeast, trypanosomes and mammalian cells.Mitochondria play vital roles in the life and death of virtually all eukaryotic cells, mediating a diverse set of functions ranging from energy production to programmed cell death. Mitochondria are also incredibly dynamic, constantly moving, fusing and dividing. During cell growth, additional mitochondria are assembled from the import of proteins and lipids from the cytosol. In three very different systems we are investigating the molecular basis of these fascinating activities.
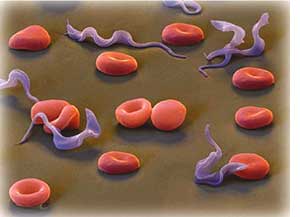
Mitochondria and kDNA in Trypansosomes. Trypanosomes, the parasites that cause important tropical diseases like African sleeping sickness, have very distinct mitochondrial functions and morphologies when they propagate within the insect vector, compared to when they infect their mammalian host. In collaboration with Dr. Paul Englund (Department of Biological Chemistry), we are studying how mitochondria are remodeled during the trypanosome life cycle. The mitochondrial DNA of trypanosomes (called a kinetoplast or kDNA) is very unique, consisting of several thousand DNA rings topologically intertwined into a giant DNA network. We are also examining the novel mechanisms by which kinetoplast DNA replicates, and how the interlocked kDNA is segregated to the two daughter cells during cytokinesis. Besides presenting a fascinating paradigm for cell biology research, the unusual mitochondria and kDNA of trypanosomes provide attractive drug targets.
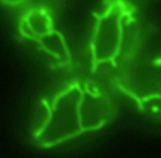
Mitochondrial Dynamics in Yeast. Most mitochondrial proteins are encoded on nuclear genes, synthesized in the cytoplasm, and imported into the organelle via a multi-step pathway that includes binding to surface receptors on the mitochondrial outer membrane, unfolding of the precursor, translocation across both mitochondrial membranes via large protein-conducting channels, and refolding and assembly of the imported protein inside the organelle. Using the tools of yeast molecular genetics, we have identified much of the machinery that mediates the mitochondrial protein import. Using a variety of different techniques, we are now probing the molecular mechanisms by which this machinery functions.
Compared to proteins imported into mitochondria, much less is known about how mitochondria divide, how mitochondria fuse, how the organelles are transmitted to daughter cells during cell division, or how mitochondria structure is established and maintained. To address these questions, we have isolated and are analyzing yeast mutants defective in the number, the location and the shape of their mitochondria.

Mitochondrial dysfunction in human disease. Because mitochondrial function intersects so many different metabolic pathways in the cell, it is not surprising that many diseases are attributable to dysfunction of the organelle. In collaboration with Dr. Richard Kelley (Kennedy-Kreiger Institute), we are studying a mitochondrial disease, called DCMA (dilated cardiomyopathy with ataxia) characterized by early-onset heart problems, growth retardation, loss of neutrophils, and uncoordinated movement. We find that DCMA patients carry mutations in a component of the human mitochondrial protein import machinery. Strikingly, DCMA patients have virtually identical problems as those with Barth Syndrome (BTHS), caused by a defect in the production of a mitochondrial-specific lipid. We speculate that both diseases impact a common step within the mitochondria, and we are actively tracking down the cellular defects shared in cells lines established from different patients.